Wanneer is registratie van een medicijn (g)een optie?
Registratie (oftewel een handelsvergunning) is een optie zodra volgens het College ter Beoordeling van Geneesmiddelen (CBG) aan alle voorwaarden (effectiviteit, kwaliteit, veiligheid) is voldaan. In principe kan en mag iedereen een medicijn laten registeren. Meestal wordt het gedaan door commerciële partijen, maar een ziekenhuis of een not-for-profitorganisatie kan ook een medicijn registreren.
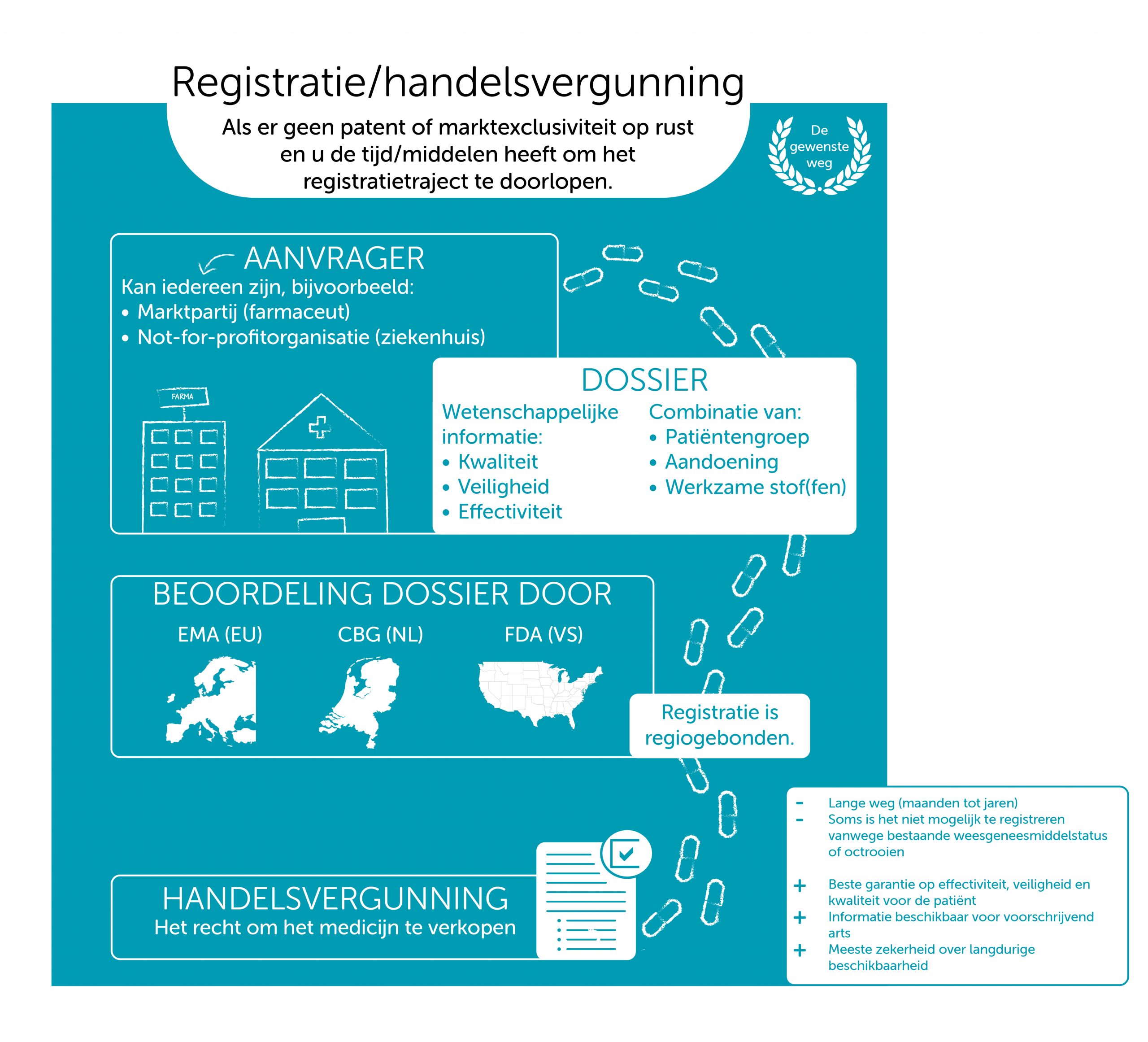
Een registratie is geen optie als er nog intellectueel eigendom op het geneesmiddel zit, zoals een patent op de werkzame stof of op een essentieel onderdeel van het productieproces. (NB: strikt genomen is registratie dan wel mogelijk, maar zinloos omdat er een verkoopverbod op het middel geldt.)
Het registreren van een concurrerend middel is ook geen optie wanneer er een medicijn op de markt is voor dezelfde indicatie met een actieve weesgeneesmiddelenstatus. Tien jaar na het op de markt komen van een medicijnen met een actieve weesgeneesmiddelenstatus wordt er geen vergunning afgegeven voor eenzelfde medicijn voor dezelfde ziekte (theoretisch kan de houder van de weesgeneesmiddelenstatus daar wel toestemming voor geven). Dat betekent in de praktijk dat de aanbieder/producent van het middel tien jaar lang de enige is die het middel mag verkopen in de Europese Unie (we spreken dan van marktexclusiviteit).
Voor een andere indicatie zou het dan wel geregistreerd mogen worden, maar dat biedt geen soelaas voor de zeldzame indicatie, omdat het medicijn alleen mag worden voorgeschreven voor de indicatie waarvoor deze geregistreerd is.
Zelf een geneesmiddel registreren
Kiezen tussen nationaal of EU
Wie zelf een medicijn wil registreren, kan kiezen tussen een nationale of een EU-vergunning. Die keuze vervalt als er een weesgeneesmiddelstatus (orphan drug designation) in het spel is. Als deze status is toegekend, is een EU-registratie de enige weg. Voor sommige andere specifieke gevallen, bijvoorbeeld biologicals en specifieke ziektegebieden, is de EU-registratie ook de enige mogelijkheid. Het vergunningstraject voor de EU wordt de Europese route of de centrale route genoemd.
Een vergunning voor de EU wordt verleend door Europese Commissie op voordracht van de Committee for Medicinal Products for Human Use (CHMP) van de European Medicines Agency (EMA).
Van nationale procedure naar andere EU-lidstaten
In Nederland wordt de nationale vergunning verleend door het College ter Beoordeling van Geneesmiddelen (CBG). Na een nationale procedure is de vergunning alleen in een specifiek land geldig.
De vergunninghouder kan vervolgens via een wederzijdse erkenningsprocedure vergunningen aanvragen voor andere EU-landen. Via de decentrale procedure kan een vergunning worden aangevraagd in meerdere Europese landen tegelijk. Er is dan één land dat optreedt als ‘beoordelaar’.
Dossier indienen voor de beoordelingsprocedure van de registratie/vergunning
In de beoordelingsprocedures voor de vergunning wordt gekeken naar veiligheid, effectiviteit en kwaliteit van het medicijn. De aanvrager moet daarover een dossier indienen. Dit document (het Common technical document, kortweg CTD) heeft een vaste opbouw van vijf modules.
Module 1 bevat administratieve data (gegevens producten) en de Summary of Product Characteristics (SmPC), oftewel de bijsluiter en wat er op de verpakking moet staat. De SmPC bevat de belangrijkste wetenschappelijke informatie over het medicijn voor artsen en apothekers.
Module 2 is de samenvatting van modules 3, 4 en 5: verkorte weergaven van de chemisch-farmaceutische, farmacologisch-toxicologisch en klinisch-farmacologische beschrijvingen.
Module 3 bevat de chemisch-farmaceutische gegevens. Dat zijn alle gegevens over de samenstelling en bereiding van het medicijn en de gegevens van de kwaliteitscontroles.
Module 4 bevat de farmacologische toxicologische gegevens. Hierin staat alle informatie over een medicijn uit preklinische onderzoeken (in vitro en in dieren), gerelateerd aan de toxiciteit en het werkingsmechanisme.
Module 5 bevat de klinisch farmacologische gegevens. Daarin staan de gegevens over effectiviteit en veiligheid voor mensen.
Voor een nieuw medicijn moet een volledig dossier (full application) worden ingediend. Dat is tijdrovend en kostbaar. Voor medicijnen waarover al meer bekend is hoeft de aanvrager niet alle informatie zelf aan te leveren en zijn aangepaste procedures mogelijk (bijvoorbeeld generic, hybrid of biosimilar applications). De volgende aanvraagprocedures worden onderscheiden:
1. Full application – Article 8(3)
2.
a) Generic, Hybrid or similar biological applications – Article 10
b) Well-established use application – Article 10a
c) Fixed combination application – Article 10b
d) Informed consent application – Article 10c
e) A generic of a reference medicinal product – Article 10.1
f) Hybrid application – Article 10.3 Biosimilar application – Article 10.4
Voor elk van deze procedures gelden andere eisen. Zo kunnen sommige procedures sneller verlopen omdat er bijvoorbeeld geen preklinisch en/of klinisch onderzoek meer nodig is, omdat alle informatie al in de literatuur is te vinden is, of omdat er kan worden verwezen naar marktvergunningen voor vergelijkbare of dezelfde geneesmiddelen.
Voor al deze procedures moet je als indiener ook een som geld betalen. Voor de centrale procedure van de EMA is die het duurste: € 266.850 (inclusief 10% korting voor een weesgeneesmiddel). Voor de andere routes staan de prijzen vermeld bij de nationale organisatie.